

The situation around the bilateral agreements and the MRA (Mutual Recognition Agreement) between the EU and Switzerland is getting worse. Swiss manufacturers are now under enormous pressure as time runs out on their products’ certification.
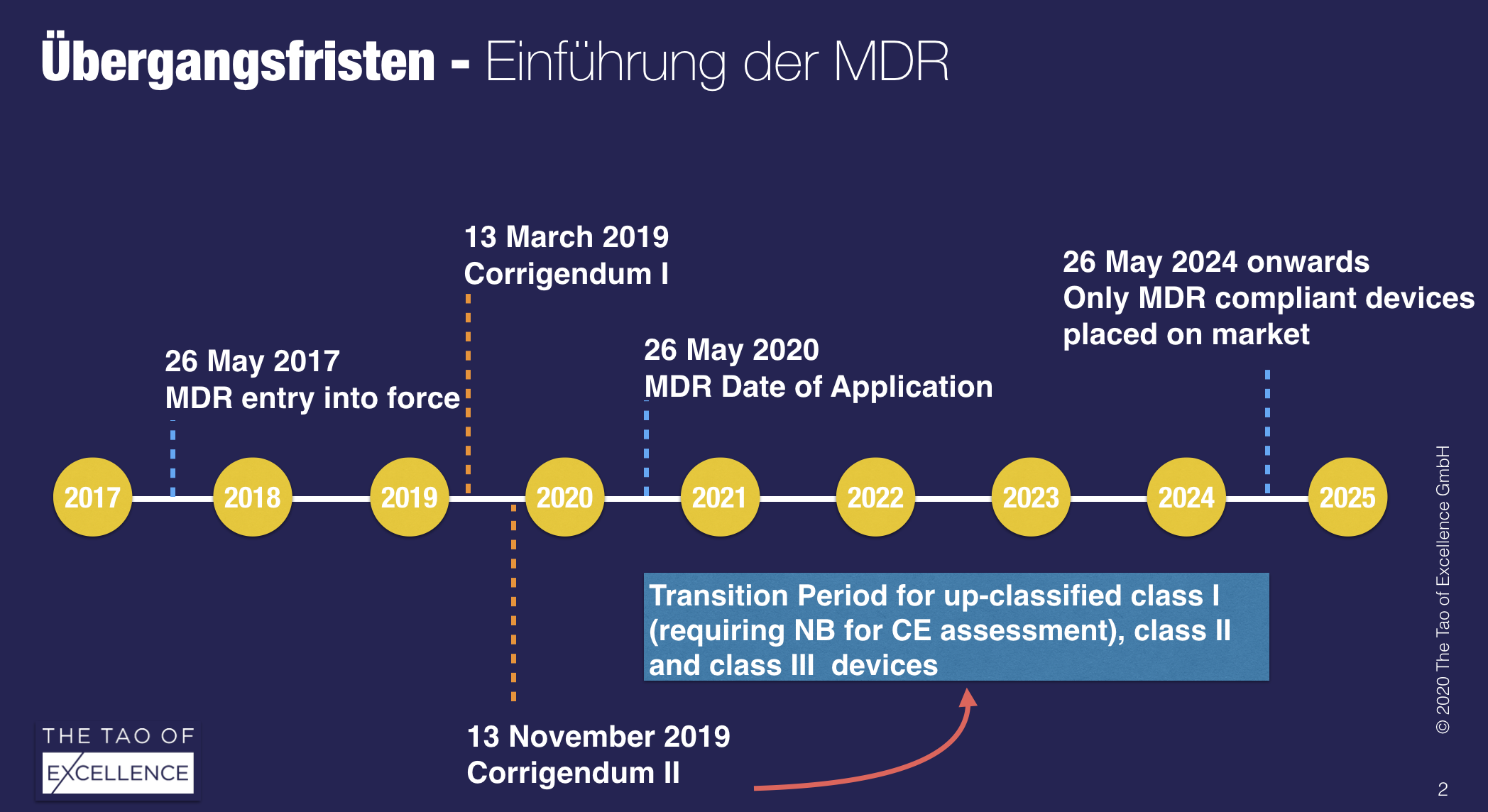
According to the latest information, it will not be possible to renew the MRA before the EU’s new Medical Device Regulation (MDR) becomes applicable from 26 May 2020, effectively barring Swiss medtech products from accessing the EU market.
The following articles have been published by the Swiss press and describe the situation as it stands:
- https://www.nzz.ch/schweiz/schweizer-medizinaltechnik-eu-erhoeht-den-druck-ld.1534093
- https://www.tagesanzeiger.ch/schweiz/standard/am-wef-soll-der-bundesrat-die-medtechbranche-retten/story/26281637
Possible scenarios
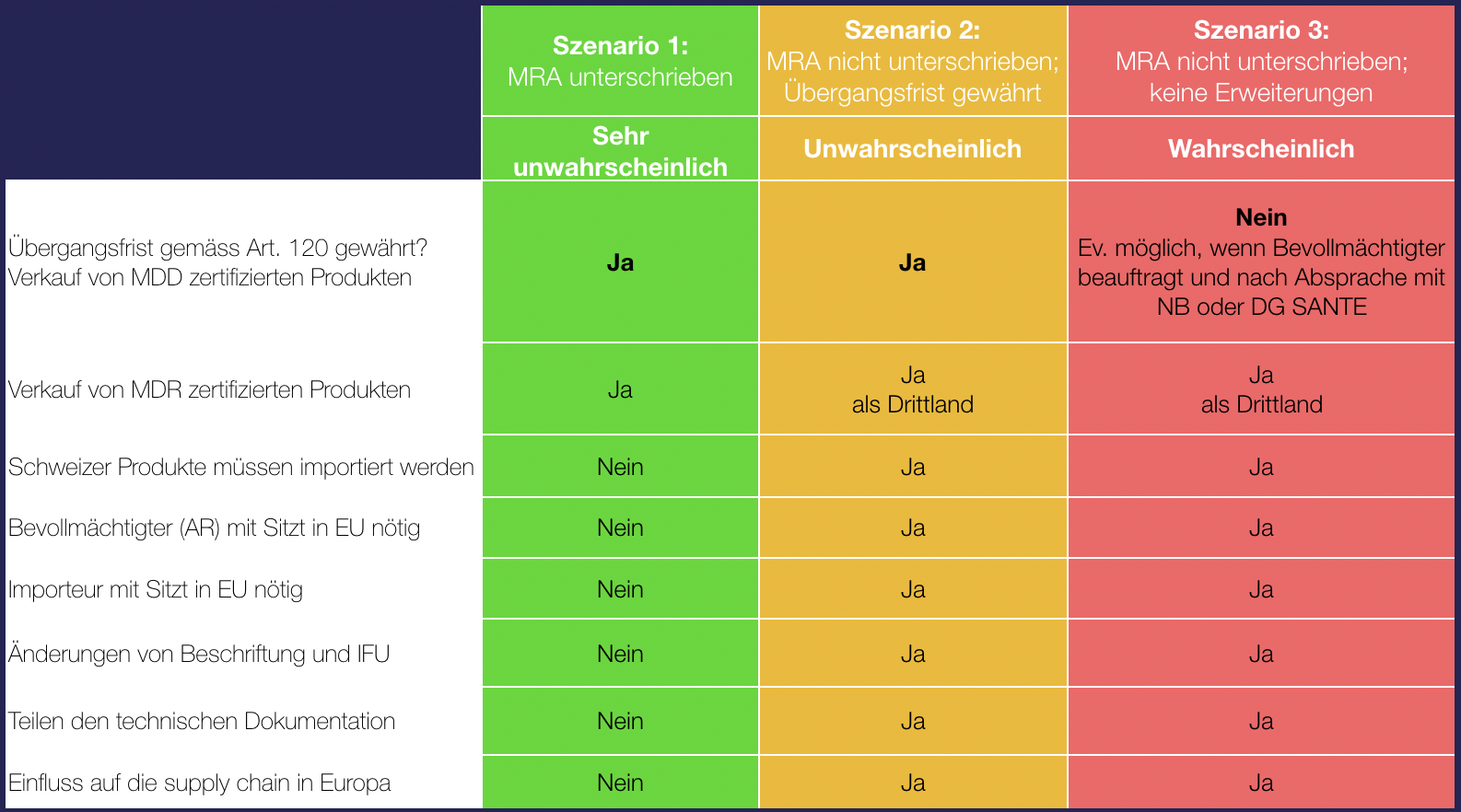
Swiss Medtech has published the possible scenarios for manufacturers:
The feared worst-case scenario is now the most likely. This means that there will be no transition period to the MDR for medical devices from Swiss manufacturers and that their MDD-certified products may no longer be sold in the EU. The situation is not mitigated by the corrigendum II for Swiss manufacturers.
We have therefore put together the following information for you:
You will be affected if you answer ‘yes’ to the following questions.
- Are you a manufacturer of medical devices according to MDR?
- Are you a Swiss distributor?
- Do you sell medical devices to Europe?
- Is the European market business relevant for you?
What has to be done now?
Get active! Prepare for scenario 3 and you will have the best starting position. Appoint a proxy in the EU before May 26, 2020.
- Re-evaluate your portfolio from a business perspective.
- Evaluate and adjust your supply chain in the EU.
- Work with your distributor to see if he has the role of importer or authorized representative. If not, you will need to find an authorized representative in the EU.
- Discuss with your Notified Body whether they agree with the transition period by the authorized representative.
- Establish all the obligations of the authorized representative and agree on the contract.
- Plan your inventory of MDD / MDR certified devices. Attention: In the worst case, no MDD / MDR device may be placed on the European market from May 27, 2020.
- Plan to include the name and address of the authorized representative on the label and IFU.
- Define the process of regularly updating and distributing the copy of the technical documentation to the authorized representative.
- Set up the process for communicating with the authorized representative:
- Request from authorities.
- Complaints and reports from health professionals and patients.
- Corrective and preventive measures to reduce the risks from the device.
Contact us directly with your questions or to find out what the best strategy is for your business.
 Jasminka Roth
Jasminka Roth
Founder and Director
Phone
+41 52 685 51 65
Email
meetus@taoexcellence.ch
 Deepa Rajagopalan
Deepa Rajagopalan
Quality Implementation Manager
Phone
+41 52 685 51 65
Email
meetus@taoexcellence.ch
Did you like this article? Follow us on LinkedIn and Twitter!